Your privacy is very important to us. When you visit our website, please agree to the use of all cookies. For more information about personal data processing, please go to Privacy Policy.




What is MECP2 Duplication Syndrome?
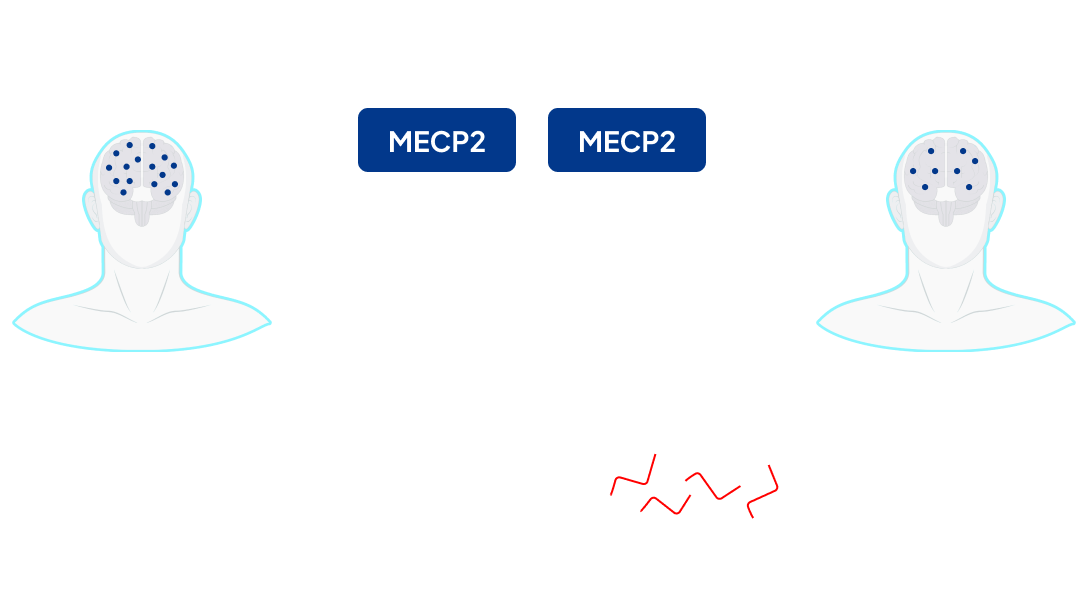
MECP2 Duplication Syndrome (MDS) is a rare, fatal and congenital-neurodevelopmental disorder, primarily affecting boys, that is caused by genomic duplications of the Xq28 region containing MECP21. MECP2 encodes for methyl-CpG-binding protein 2 (MeCP2), an epigenetic regulator that is crucial for normal brain development and maintenance of brain function2. Additional copies of the MECP2 gene lead to excess MeCP2 production, which further leads to abnormal neuronal function. There are about 50,000 to 100,000 MDS patients worldwide2-5. Currently only 50% of those diagnosed will survive past the age of 256.
1 Sarika U. Peters et al,. Phenotypic features in MECP2 duplication syndrome: Effects of age. Am J Med Genet A. 2021 Feb;185(2):362-369.
2 Yingyao Shao et al,. Antisense oligonucleotide therapy in a humanized mouse model of MECP2 duplication syndrome. Sci Transl Med. 2021 Mar 3;13(583):eaaz7785.
3 Peter Giudice-Nairn et al,. The incidence, prevalence and clinical features of MECP2 duplication syndrome in Australian children. J Paediatr Child Health. 2019 Nov;55(11):1315-1322.
4 Pallab K Maulik et al,. Prevalence of intellectual disability: a meta-analysis of population-based studies. Res Dev Disabil. 2011 Mar-Apr;32(2):419-36.
5 G.E. Utine et al,. Searching for Copy Number Changes in Nonsyndromic X-Linked Intellectual Disability. Mol Syndromol. 2012 Jan;2(2):64-71.
6 MECP2 Duplication Foundation, https://mecp2d.org/.
-
50,000 to 100,000MDS patients worldwide2-5

What Are the Symptoms Associated with MDS?
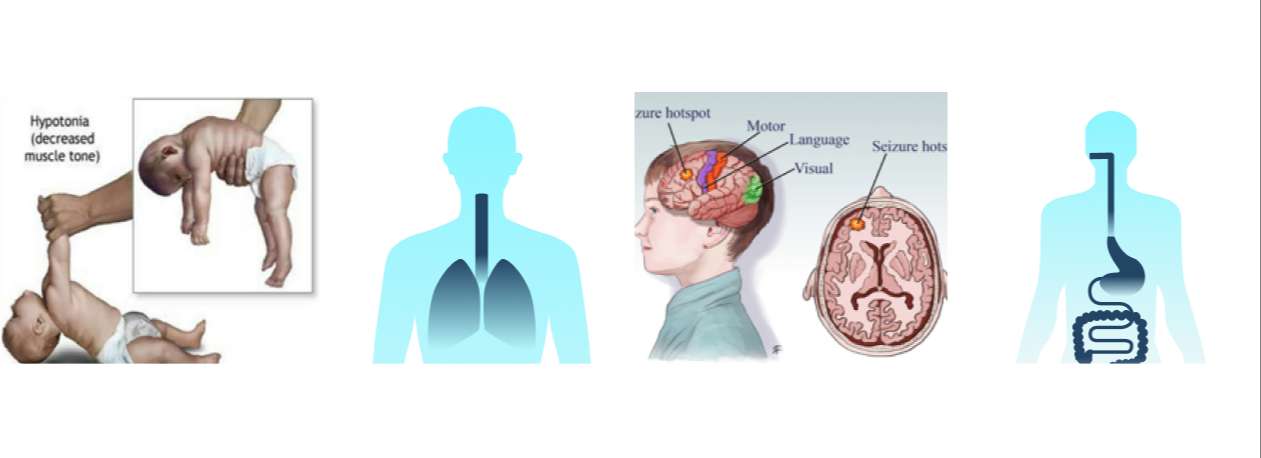
The most common clinical features of MDS include severe developmental delay, motor impairments, absent or little speech, severe mental retardation, recurrent respiratory infections, refractory epilepsy1, and shortend lifespan, with death often occurring before the age of 25 years. Other variably presenting features include autistic features, gastrointestinal dysfunction, and mild facial dysmorphism2.
1 Muharrem Ak et al,. Exploring the characteristics and most bothersome symptoms in MECP2 duplication syndrome to pave the path toward developing parent-oriented outcome measures. Mol Genet Genomic Med. 2022 Aug;10(8):e1989. 2 Van Esch H. MECP2 Duplication Syndrome. 2008 Jan 18 [updated 2020 May 21]
How is MDS Diagnosed?
The diagnosis of MECP2 duplication syndrome is established in an individual by identification of a heterozygous whole-gene duplication of MECP2 on molecular genetic testing. Molecular genetic testing in a child with developmental delay typically begins with chromosomal microarray analysis (CMA). CMA uses oligonucleotide or SNP arrays to detect genome-wide changes1. Many diagnostic laboratories now offer clinical testing for MECP2 duplication using quantitative real-time PCR, MLPA, and/or array-CGH methodologies2.

1 Van Esch H. MECP2 Duplication Syndrome. 2008 Jan 18 [updated 2020 May 21] .
2 Melissa B. Ramocki at al,. The MECP2 duplication syndrome. Am J Med Genet A. 2010 May;152A(5):1079-88.

What is the Treatment Option for MDS?
-
There is currently no treatments available for MECP2 duplication syndrome, although investigational drugs are currently in development. As of today, clinical treatment mainly focuses on relieving symptoms. The candidate therapeutic, HG204, is developed using HuidaGene's proprietary RNA editor delivered by a single AAV vector, which induces the degradation of MECP2 RNA for the treatment of MDS through a single administration. HG204 is currently in preclinical studies with the expectation of dosing patients through the investigator-initiated trial in China soon.
Previous : HG302(DMD)
Next : HG202(nAMD)